价格:410
货号2×HRbio™ Seamless Assembly and Cloning kit无缝克隆试剂盒(单/多片段连接)
产品详情
产品详情
产品信息
|
产品名称 |
产品编号 |
规格 |
|
2×HRbio™ Seamless Assembly and Cloning kit 无缝克隆试剂盒(单/多片段连接) |
HRF0112 |
10 T |
产品描述
本制品不依赖于T4连接酶,不受载体和目的片段的酶切位点限制,而直接用重叠片段重组的方法,采用特殊的酶组合可以将任意方法线性化后的载体和与其两端具有15-25bp重叠区域的PCR片段(平端)定向重组,可以快速实现1-5个片段的高效无缝克隆。
产品特点
1.15分钟可以将一个或者多个长、短PCR扩增片段(平端)插入载体。
2.不受载体和插入片段酶切位点的可用性和载体平端/粘性末端的限制,可以在任意位点进行克隆。
3.无缝克隆,插入点不会引入不需要的碱基序列。
4.高效、准确,阳性率>95% 。
运输和保存方法
干冰运输。-20℃保存,避免反复冻融。
注意事项
1) 为了您的安全和健康,请穿实验服并戴一次性手套操作。
2)本产品仅作科研用途!
原理示意图

线性化载体和插入DNA片段的制备
A:线性化载体的制备
(1)酶切来源:酶切所得线性载体,平末端或者粘端、单酶切或者双酶切均可,酶切后胶回收。
注: 无缝拼接和克隆反应体系内无双链DNA连接酶,不会发生载体自连反应。因此,即使是以单酶切方式制备的线性化载体也无需进行末端脱磷酸处理。重组产物转化后出现的假阳性克隆 (无插入片段) 是由酶切不完全未线性化环状载体转化而形成的。我们推荐酶切后胶回收可以把这种未线性化载体比例降低到最低程度。
(2)反向PCR来源:建议使用高保真DNA聚合酶制备,如果扩增条带单一可以通过PCR产物纯化获得载体,否则通过胶回收获得载体。
注:反向PCR的质粒模板也是非线性化载体,也可能导致假阳性克隆(无插入片段),因此PCR来源线性载体(PCR产物)纯化前用Dpn I内切酶消化质粒模板,可以降低背景,提高阳性率。但是一般情况下,经过胶回收已经足以把这种未线性化载体比例降低到最低,因此反向PCR来源的线性化载体我们也推荐胶回收。
B:插入DNA片段的制备
单个插入片段克隆引物设计:克隆引物包括插入片段特异性引物序列和重叠序列。
克隆正向引物(5’-3’):线性载体正向16-25 nt重叠区序列(3’末端算起)+插入片段正向特异引物序列(18-25 nt)
克隆反向引物(5’-3’):线性载体反向16-25 nt重叠区序列(3’末端算起)+插入片段反向特异引物序列(18-25 nt)
注意:重叠区的碱基数至少16 bp,并且多段重叠区域之间的Tm值需保持一致且﹥60°C (AT pair = 2°C and GC pair = 4°C),否则可延长碱基数目直到符合要求。
按照线性载体末端的结构(5’突出,3’突出,平末端),引物设计也分3种情况,示意如下:
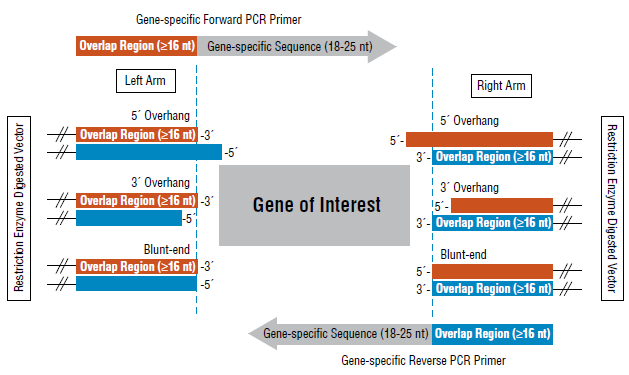
线性化载体的两端因线性方式(如单酶切、双酶切、反向PCR)不同,可以是以上三种末端结构的两两任意组合,插入片段特异性引物设计的原则遵循一般引物设计的原则即可。
计算扩增引物退火温度时,只需计算基因特异性扩增序列的Tm值,载体末端同源序列不应参与计算。
正向引物设计举例说明(EcoR I 酶切开)

如上图,载体由Ecor I 酶切开,形成5’突出末端:
根据上述设计原则,从3’端开始计算,往回算16bp-25bp(本举例采用了20 bp末端重叠相同序列),加到目的片段特异引物序列前面即可。确保重叠区域的Tm值保持一致且﹥60°C (AT pair = 2°C and GC pair = 4°C)。
正向引物具体如下:5'—GAGCCTAGGTGAGTTGGCCGNNNNNNNNNNNNNNNNNNNN—3'
注意:以上引物设计完成克隆连接后,EcoR I 酶切位点将会消失(不保留酶切位点)。
如果需要保留 EcoR I 酶切位点,需要在载体末端20 bp重叠序列和目的片段特异引物序列之间补齐缺失的EcoR I 识别位点序列aattc,完成克隆连接后,EcoR I 酶切位点依然存在(保留酶切位点)。
具体如下:5' — GAGCCTAGGTGAGTTGGCCG aattc NNNNNNNNNNNNNNNNNNNN —3'
反向引物设计举例说明(Hind III 酶切开)

如上图,载体由Hind III 酶切开,形成5’突出末端:
根据上述设计原则,从3’端开始计算,往回算16bp-25bp(本举例采用了20 bp末端重叠相同序列),加到目的片段特异引物序列前面即可。确保重叠区域的Tm值保持一致且﹥60°C (AT pair = 2°C and GC pair = 4°C)。
反向引物具体如下:5' —CTGCCATCGGATCGTTCGCANNNNNNNNNNNNNNNNNNNN—3'
注意:以上引物设计完成克隆连接后,Hind III 切位点将会消失(不保留酶切位点)。
如果需要保留 Hind III 酶切位点,需要在载体末端20 bp重叠序列和目的片段特异引物序列之间补齐缺失的Hind III 识别位点序列agctt ,Hind III 酶切位点依然存在(保留酶切位点)。
具体如下:5' — CTGCCATCGGATCGTTCGCA agctt NNNNNNNNNNNNNNNNNNNN —3'
(2) 多个插入片段的克隆引物设计:与载体两端连接的插入片段引物设计方法同单片段设计方法,片段与片段之间连接的插入片段引物设计方法见下图例:
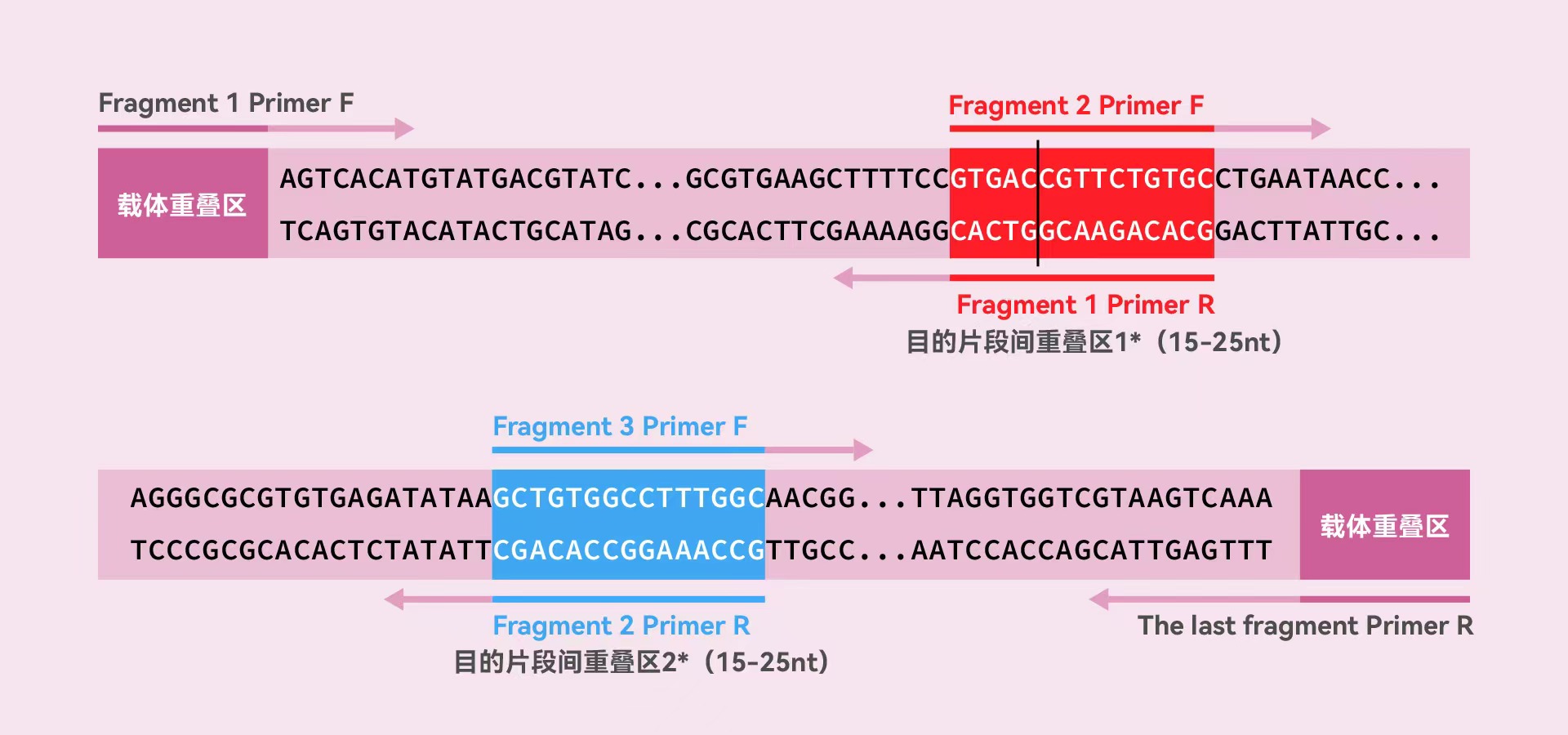
* 多个插入片段之间重叠区设计有上述*标记(蓝色和红色)的两种方式,在多片段引物设计时,选择任意一种方式或者两种方式混用均可,保证片段与片段之间有15-25bp 的重叠区。
(3) 酶的选择:建议使用高保真DNA聚合酶或者PCR Mix。
(4) 反应条件:一般按照具体使用的聚合酶说明书进行即可。
(5) 纯化插入片段
可选:如果片段来源于质粒模板,且该质粒与重组载体具有相同抗性,纯化前用Dpn I内切酶消化质粒模板,可降低背景,提高阳性率。
如果片段单一,则建议用PCR产物纯化试剂盒纯化片段。
如果有非特异扩增,则建议用胶回收试剂盒回收片段。
使用该方法克隆,用来线性化载体的酶切位点在拼接的时候会缺失,如果对酶切位点有严格要求的,建议注意酶切位点的择,必要时可在正反向克隆引物的重叠区序列和特异基因序列之间增加缺失掉的碱基来恢复原有酶切位点(见前述正反向引物设计举例说明)。
如果重组质粒用于蛋白表达,则在引物设计时注意读码框,蛋白表达及纯化所需序列(如启动子,RBS序列,起始密码子,终止密码子,蛋白标签等)不被破坏。
无缝克隆反应的操作步骤
注意:2×Cloning Master Mix 含有连接增强剂PEG很粘稠,从冰箱拿出来温度低时更粘稠,可以放在手心化冻几分钟提高温度便可降低粘稠度(不影响质量),手指拨打离心管底充分混匀或者涡旋震荡混匀。
1.按照下表建立反应体系(可使用PCR管在室温配制)
|
2 × Cloning Master Mix |
5 μL |
|
Linear Vector (10-80 ng) |
X μL* |
|
Insert(s) |
Y μL* |
|
dd H2O |
To 10 μL |
* 载体一般用10-50 ng,插入片段与载体的摩尔比在2:1-3:1之间最佳;如果插入片段小于200bp,插入片段与载体的摩尔比用5:1,多片段连接的情况下,片段与片段之间摩尔比为1:1。
2.轻轻吹打混匀,在50°C反应15分钟(可在PCR仪器上进行),反应结束后,将PCR管置冰上 后直接转化或者保存于-20°C。较短片段例如100bp-1kb只需要15分钟便能获得足够转化子,较长片段的连接,可以延长反应时间到30-60分钟。超过3个片段的连接,可以延长反应时间到60分钟。
3.取5 μL 反应产物按照感受态细胞说明书进行转化(如果转化子较少,可以将所有的产物转化并将所有的转化液涂板)。
阳性克隆的鉴定
可根据具体情况,选择菌落PCR鉴定,提取质粒进行限制性内切酶鉴定或测序鉴定。